
Protein dari bakteri Staphylococcus aureus
Pada akhir November, tim DeepMind Google mengumumkan bahwa sistem pembelajaran mendalam AlphaFold telah mencapai tingkat akurasi yang belum pernah terjadi sebelumnya dalam memecahkan masalah pelipatan protein , masalah yang sulit dalam biokimia komputasi.
Apa masalahnya dan mengapa begitu sulit untuk dipecahkan?
Protein adalah rantai panjang asam amino. Kode DNA Anda untuk urutan ini, dan RNA membantu membuat protein sesuai dengan cetak biru genetik ini. Protein disintesis dalam bentuk rantai linier, tetapi kemudian dilipat menjadi struktur bola yang kompleks (lihat gambar di awal artikel).
Sebagian rantai bisa menggulung menjadi spiral yang rapat, " α-helix . "Bagian lain dapat menekuk ke depan dan ke belakang untuk membentuk gambar datar yang lebar," β-sheet ":
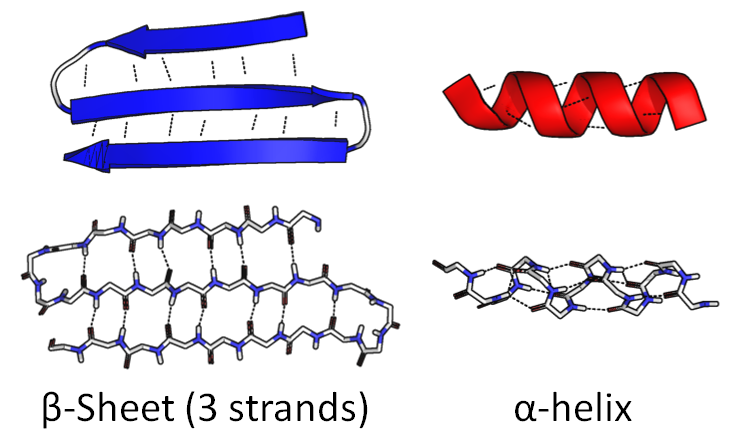
Urutan asam amino itu sendiri disebut struktur primer . Gambar-gambar ini disebut struktur sekunder .
Komponen ini sendiri juga terlipat untuk membentuk bentuk kompleks yang unik. Ini disebut struktur tersier :
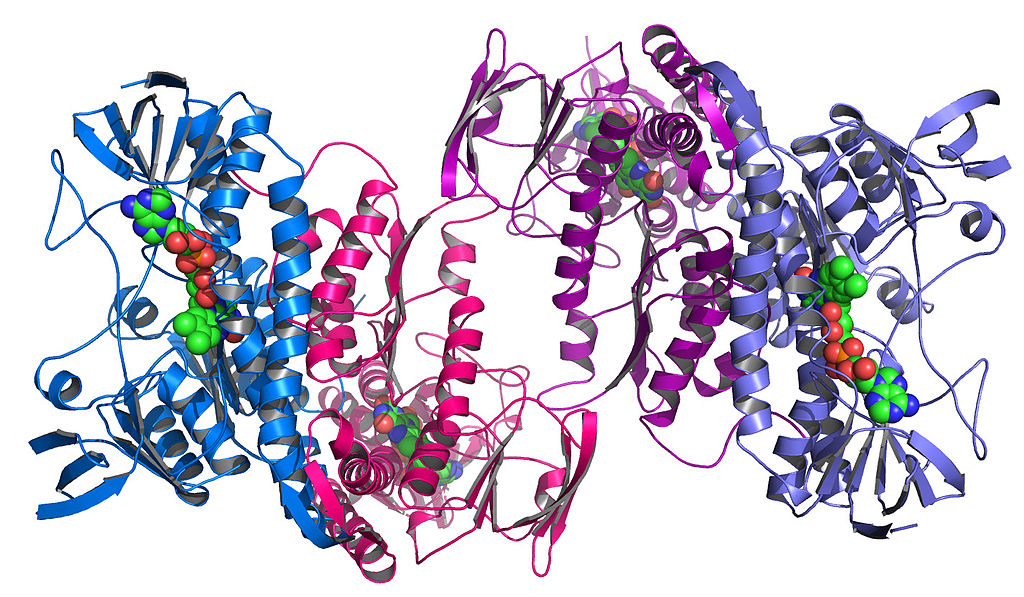
Enzim yang diambil dari bakteri Colwellia psychrerythraea

protein RRM3
Terlihat berantakan. Mengapa bola asam amino yang kusut ini begitu penting?
Struktur protein tidak acak! Setiap protein terlipat menjadi struktur yang berbeda, unik, dan sebagian besar dapat diprediksi, yang penting agar dapat berfungsi dengan baik. Karena bentuk fisiknya, protein sangat cocok dengan struktur yang dapat diikatnya. Sifat fisik lainnya juga penting, terutama distribusi muatan listrik di atas protein. Pada gambar, muatan positif ditunjukkan dengan warna biru, muatan negatif berwarna merah:

Distribusi muatan permukaan pada protein pembawa lipid tanaman 1 beras
Jika protein pada dasarnya adalah mesin nano yang merakit sendiri, maka tujuan utama urutan asam amino adalah untuk menghasilkan bentuk uniknya, distribusi muatan, dan segala sesuatu yang menentukan fungsi protein. Bagaimana tepatnya proses ini terjadi belum sepenuhnya jelas - sekarang ini adalah bidang penelitian yang aktif.
Bagaimanapun, memahami struktur itu penting untuk memahami cara kerjanya. Namun, urutan DNA hanya menentukan struktur utama protein. Bagaimana kita mengetahui struktur sekunder dan tersiernya - yaitu, bentuk persis dari jalinan ini?
Masalah ini disebut masalah pelipatan protein, dan ada dua pendekatan dasarnya: pengukuran dan prediksi.
Metode eksperimental dapat mengukur struktur suatu protein. Namun, ini tidak mudah dilakukan: struktur tidak terlihat melalui mikroskop optik. Untuk waktu yang lama, kristalografi sinar-X adalah metode utama untuk mempelajari struktur. Selain itu, resonansi magnetis nuklir digunakan, dan baru-baru ini muncul teknologi baru, mikroskop cryoelectron .

Pola difraksi sinar-X protease SARS
Namun, metode ini mahal, rumit dan memakan waktu, dan selain itu, metode ini tidak bekerja dengan semua protein. Secara khusus, protein yang tertanam dalam membran sel - reseptor angiotensin converting enzyme 2 (ACE2) yang sama yang mengikat virus COVID-19 - terlipat ke dalam lapisan ganda lipid. sel, dan sangat sulit untuk mengkristal.

Struktur membran sel
Oleh karena itu, kami dapat membongkar struktur sebagian kecil dari protein yang diurutkan . Basis data protein universal berisi 180 juta urutan, sedangkan basis data struktur protein tiga dimensi hanya memuat 170 ribu posisi.
Kami membutuhkan metode yang lebih baik.
* * *
Ingatlah bahwa struktur protein sekunder dan tersier pada dasarnya adalah fungsi dari struktur primer yang kita kenal melalui pengurutan. Bagaimana jika, alih-alih mengukur struktur protein, kita dapat memprediksinya?
Ini adalah tugas untuk memprediksi struktur protein. Ahli biokimia komputasi telah mengerjakannya selama beberapa dekade.
Bagaimana Anda bisa mendekatinya?
Cara yang jelas adalah dengan mensimulasikan fisika proses secara langsung. Kami mensimulasikan gaya untuk setiap atom, dengan mempertimbangkan lokasi, muatan, dan ikatan kimianya. Kami menghitung akselerasi dan kecepatan, dan langkah demi langkah menelusuri evolusi sistem. Ini disebut "dinamika molekuler".

Superkomputer " Anton " oleh DE Shaw Research

Superkomputer IBM Blue Gene
Online puzzle Foldit
Masalahnya adalah bahwa pendekatan ini sangat intensif secara komputasi. Protein tipikal mengandung ratusan asam amino, yaitu ribuan atom. Lingkungan juga penting: saat melipat, protein berinteraksi dengan air di sekitarnya. Oleh karena itu, perlu dilakukan simulasi perilaku sekitar 30 ribu atom. Dalam hal ini, interaksi elektrostatis terjadi antara setiap pasangan atom, dengan perkiraan kasar, kita mendapatkan 450 juta pasang, masalah dengan kompleksitas O (N2). Ada algoritma pintar yang mengurangi kompleksitasnya menjadi O (N log N). Selain itu, untuk simulasi, perlu untuk menghitung 10 9 -10 12 langkah. Sakit kepala yang luar biasa.
Oke, tapi kita tidak perlu mensimulasikan seluruh proses pelipatan. Pendekatan lain menyarankan untuk menemukan struktur dengan energi potensial minimal. Objek biasanya cenderung berhenti dengan energi paling sedikit, jadi pendekatan heuristik ini dibenarkan. Energi dapat dihitung dengan model dinamika molekul yang sama, yang memberikan kita besarnya interaksi. Dengan pendekatan ini, kita dapat mencoba sekelompok kandidat dan memilih struktur dengan energi paling sedikit. Masalahnya, tentu saja, dari mana mendapatkan struktur itu. Jumlahnya terlalu banyak - ahli biologi molekuler Cyrus Levintol telah menghitung bahwa mungkin ada sekitar 10.300 . Secara alami, Anda dapat menggunakan pendekatan yang lebih cerdas daripada kekerasan acak. Tapi jumlahnya masih terlalu banyak.
Oleh karena itu, banyak upaya telah dilakukan untuk mempercepat perhitungan tersebut. Anton, seorang superkomputer dari DE Shaw Research, menggunakan peralatan khusus - sirkuit terintegrasi khusus. IBM juga menggunakan superkomputer bio Blue Gene. Stanford meluncurkan proyek Folding @ Home, menggunakan daya komputer rumah yang didistribusikan. Proyek Foldit UW mengubah melipat menjadi permainan untuk menambahkan intuisi manusia pada komputasi.
Namun, untuk waktu yang lama, belum ada teknologi yang mampu memprediksi berbagai macam struktur protein dengan akurasi tinggi. Pada kompetisi CASP yang diadakan dua kali setahun, dimana hasil algoritma dibandingkan dengan struktur yang diukur secara eksperimental, tempat pertama menerima prediksi dengan akurasi 30-40%. Sampai saat ini:

Akurasi prediksi median tim terbaik dalam kategori pemodelan gratis.
Bagaimana cara kerja AlphaFold? Ini menggunakan beberapa jaringan saraf dalam untuk mempelajari berbagai fungsi yang terkait dengan setiap protein. Salah satu fungsi utamanya adalah memprediksi jarak yang dihasilkan antara pasangan asam amino. Ini membawa algoritme ke struktur akhir. Dalam satu varian algoritme (dijelaskan dalam jurnal Nature and Proteins ), fungsi potensial dari prediksi ini diturunkan, di mana penurunan gradien paling sederhana diterapkan, yang bekerja dengan sangat baik.
Keuntungan utama AlphaFold dibandingkan metode sebelumnya adalah tidak perlu membuat asumsi tentang struktur. Beberapa metode bekerja dengan memecah protein menjadi beberapa bagian, menghitung masing-masing, dan kemudian menyatukan semuanya. AlphaFold tidak membutuhkan ini.
Rupanya, DeepMind menganggap masalah lipat ini harus diselesaikan, yang menurut saya merupakan penyederhanaan yang berlebihan, tetapi bagaimanapun juga, kemajuan mereka signifikan. Para ahli yang tidak berafiliasi dengan Google menggunakan julukan seperti " fantastis " dan " revolusioner ".
Rekayasa genetika sekarang memiliki dua alat canggih, CRISPR dan pelipatan protein. Mungkin tahun 2020-an akan menjadi bioteknologi seperti tahun 1970-an untuk komputasi.
Selamat kepada para peneliti DeepMind atas terobosan ini!