Misalnya air:
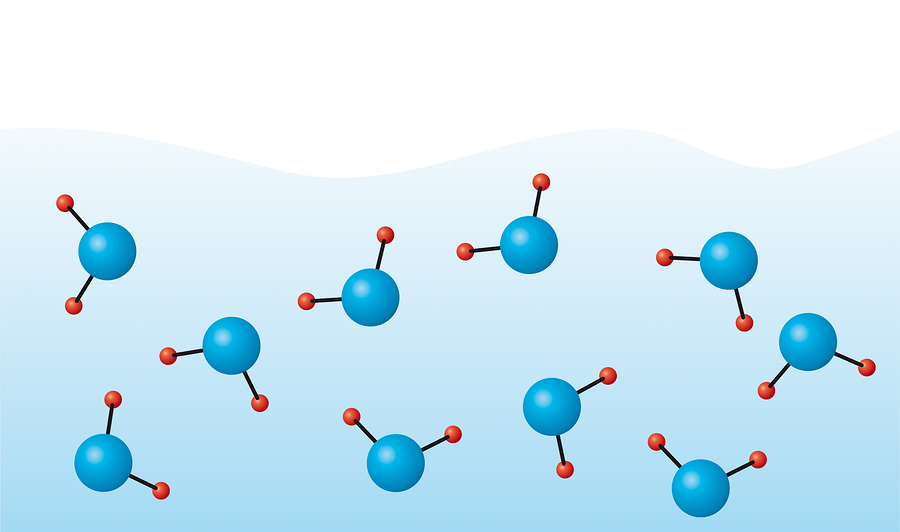
jelas bahwa hidrogen dan oksigen di dalam satu molekul berinteraksi dengan cara yang sama sekali berbeda dari oksigen yang sama dengan hidrogen di molekul tetangga. Dengan demikian, interaksi intramolekuler dan antarmolekul dibedakan. Interaksi antarmolekul dapat ditentukan oleh potensi jarak pendek dan pasangan Coulomb, yang telah dibahas di artikel sebelumnya. Di sini kita akan fokus pada intramolekuler.
Jenis interaksi intramolekuler yang paling umum adalah ikatan kimia (valensi). Ikatan kimia ditentukan oleh ketergantungan fungsional energi potensial pada jarak antara atom terikat, yaitu, oleh potensi pasangan yang sama. Namun, tidak seperti potensi pasangan biasa, interaksi ini ditentukan bukan untuk jenis partikel tertentu, tetapi untuk pasangan partikel tertentu (berdasarkan indeksnya). Bentuk fungsional yang paling umum untuk potensi ikatan kimia adalah potensial harmonik:
dimana r adalah jarak antar partikel, k adalah konstanta kekakuan ikatan, dan r 0 adalah panjang ikatan kesetimbangan; dan potensi Morse :
dimana D adalah kedalaman sumur potensial, parameter α mencirikan lebar sumur potensial.
Jenis interaksi intramolekul berikutnya adalah sudut ikatan. Mari kita lihat kembali gambar judulnya. Mengapa molekul digambarkan sebagai sudut, karena gaya elektrostatis seharusnya memberikan jarak maksimum antara ion hidrogen, yang bersesuaian dengan sudut HOH yang sama dengan 180 °? Faktanya adalah tidak semuanya tergambar dalam gambar. Dari kursus kimia sekolah, Anda dapat mengingat bahwa oksigen memiliki lebih banyak pasangan elektron bebas, yang interaksinya mendistorsi sudut:

Dalam dinamika molekuler klasik, objek seperti elektron atau awan elektron biasanya tidak dimasukkan, oleh karena itu, potensial sudut valensi digunakan untuk mensimulasikan sudut yang "benar"; ketergantungan fungsional energi potensial pada koordinat 3 partikel. Salah satu potensi yang paling nyaman adalah harmonik kosinus:
di mana θ adalah sudut yang dibentuk oleh triplet partikel, k adalah konstanta kekakuan, dan θ 0 adalah sudut kesetimbangan.
Ada potensi intramolekul dengan urutan yang lebih tinggi, misalnya, sudut torsi , tetapi lebih artifisial daripada sudut ikatan.
Menambahkan interaksi antar partikel dengan indeks yang telah ditentukan adalah hal yang sepele. Untuk tautan, kami menyimpan larik yang berisi indeks partikel tertaut dan jenis interaksi. Kami memberi setiap utas rentang tautannya sendiri untuk diproses dan Anda selesai. Begitu juga dengan sudut ikatan. Oleh karena itu, kami akan segera mempersulit tugas untuk diri kami sendiri: kami akan menambahkan kemampuan untuk membuat / menghilangkan ikatan kimia dan sudut ikatan waktu proses. Ini segera membawa kita keluar dari bidang dinamika molekuler klasik dan membuka cakrawala kemungkinan baru. Jika tidak, Anda dapat mengunduh sesuatu dari paket yang ada, misalnya LAMMPS , DL_POLY atau GROMACS , terutama karena paket tersebut didistribusikan secara gratis.
Sekarang untuk beberapa kode. Mari tambahkan bidang yang sesuai ke struktur utama:
//bonds:
int nBond; // number of bonds
int mxBond; // maximal number of bonds
int4* bonds; // array of bonds
int* nbonds; // count of bond for a given atom
int* neighToBind; // a neighbor of a given atom for binding
int* canBind; // flags that atom[iat] can be bind
int* r2Min; // distances for the nearest neighbor (used for binding)
int* parents; // indexes of one of the atom bonded with a given
cudaBond* bondTypes;
int** def_bonds; // array[nSpec][nSpec] of default bond types
int** bindBonds; // array[nSpec][nSpec] bond types created by binding
float** bindR2; // square of binding distance [nSpec][nSpec]
//angles:
int nAngle; // number of angles
int mxAngle;
int4* angles; // array of angles
int* nangles; // number of angles for given atom
int* oldTypes;
cudaAngle* angleTypes;
int* specAngles; // [nSp] angle type formed by given species
Jumlah tautan dan sudut adalah variabel, tetapi hampir selalu Anda dapat memperkirakan semaksimal mungkin dan mengalokasikan memori segera ke maksimum, agar tidak mengalokasikan memori secara berlebihan , bidang nBond dan mxBond , masing-masing, berarti jumlah tautan saat ini dan maksimum. Array obligasi akan berisi indeks atom yang akan diikat, jenis ikatan dan waktu pembentukan ikatan (jika kita tiba-tiba tertarik pada statistik seperti umur rata-rata ikatan). Array sudut akan menahan indeks triplet atom membentuk sudut ikatan dan jenis sudut ikatan. The bondTypes dan angleTypes array akan berisi karakteristik dari kemungkinan potensi ikatan dan sudut. Berikut strukturnya:
struct cudaBond
{
int type; // potential type
int spec1, spec2; // type of atoms that connected by this bond type
int new_type[2]; // bond type after mutation
int new_spec1[2], new_spec2[2];
int mxEx, mnEx; // flags: maximum or minimum of bond length exists
float p0, p1, p2, p3, p4; // potential parameters
float r2min, r2max; // square of minimal and maximal bond length
float (*force_eng)(float r2, float r, float &eng, cudaBond *bond); // return energy
int count; // quantity of such bonds
float rSumm; // summ of lentghs (for mean length calculation)
int rCount; // number of measured lengths (for mean length calculation)
int ltSumm, ltCount; // for calculation of lifetime
};
struct cudaAngle
{
int type; // potential type
float p0, p1, p2; // potential parameters
void (*force_eng)(int4* angle, cudaAngle* type, cudaMD* md, float& eng);
};
Field type mendefinisikan bentuk fungsional dari potensi, new_type , new_spec1 dan new_spec adalah indeks dari jenis ikatan dan jenis atom yang akan diikat setelah ikatan berubah (putus atau berubah menjadi jenis ikatan yang berbeda). Bidang ini direpresentasikan sebagai larik dengan dua elemen. Yang pertama sesuai dengan situasi ketika panjangnya menjadi lebih pendek dari r2mnt 1/2 , yang kedua - ketika melebihi r2max 1/2... Bagian tersulit dari algoritme adalah penerapan properti semua ikatan, dengan mempertimbangkan kemungkinan kerusakan dan transformasinya, serta fakta bahwa aliran lain dapat memutuskan ikatan tetangga, yang menyebabkan perubahan dalam jenis atom terikat. Izinkan saya menjelaskan menggunakan contoh air yang sama. Awalnya, molekulnya netral secara elektrik, ikatan kimia dibentuk oleh elektron yang umum pada hidrogen dan oksigen. Secara kasar, kita dapat mengatakan bahwa muatan pada atom hidrogen dan oksigen adalah nol (pada kenyataannya, kerapatan elektron bergeser menjadi oksigen, oleh karena itu, ada sedikit plus pada hidrogen, δ +, dan pada oksigen - 2δ-). Jika kita memutuskan ikatan, oksigen akhirnya akan mengambil elektron untuk dirinya sendiri, dan hidrogen akan melepaskannya. Partikel yang dihasilkan adalah H + dan O - . Secara total, 5 jenis partikel diperoleh, mari kita tentukan secara konvensional: H, H + , O, O- , O 2- . Yang terakhir ini terbentuk jika kita melepaskan kedua hidrogen dari molekul air. Dengan demikian, reaksi:
H 2 O -> H + + OH -
dan
OH - -> H + + O 2- .
Para ahli kimia akan mengoreksi saya bahwa dalam kondisi standar untuk air, tahap pertama dekomposisi praktis tidak dilaksanakan (dalam kesetimbangan, hanya satu molekul dari 10 7dipisahkan menjadi ion, dan bahkan kemudian tidak seperti yang tertulis). Tetapi untuk deskripsi algoritma, skema seperti itu akan bersifat ilustratif. Misalkan satu aliran memproses satu ikatan dalam molekul air, dan aliran lainnya memproses ikatan kedua dari molekul yang sama. Dan kebetulan kedua ikatan itu harus diputuskan. Kemudian satu aliran harus mengubah atom menjadi H + dan O - , dan aliran kedua menjadi H + dan O 2- . Tetapi jika aliran melakukan ini secara bersamaan, pada saat permulaan prosedur, oksigen dalam keadaan O dan kedua aliran mengubahnya menjadi O - , yang tidak benar. Bagaimanapun kita perlu mencegah situasi seperti itu. Diagram blok dari suatu fungsi yang menangani ikatan kimia:

Kami memeriksa apakah jenis atom saat ini sesuai dengan jenis koneksi, jika tidak, maka kami ambil dari tabel tipe default (harus dikompilasi terlebih dahulu), kemudian kami menentukan kuadrat jarak antar atom (r 2 ) dan, jika sambungan menyiratkan panjang maksimum atau minimum, kami memeriksa apakah itu tidak keluar apakah kita berada di luar batasan tersebut. Jika ya, maka kita perlu mengubah jenis koneksi atau menghapusnya dan dalam kedua kasus mengubah jenis atom. Untuk ini, fungsi atomicCAS akan digunakan- kami membandingkan jenis atom saat ini dengan yang seharusnya dan dalam hal ini kami menggantinya dengan yang baru. Jika jenis atom telah diubah oleh utas lain, kita kembali ke awal untuk mengganti jenis tautan. Kasus terburuk jika kita berhasil mengubah jenis atom pertama, tetapi bukan yang kedua. Sudah terlambat untuk kembali, karena setelah kita mengganti atom pertama, utas lain sudah dapat melakukan sesuatu dengannya. Apa jalan keluarnya? Saya menyarankan agar kita berpura-pura bahwa kita memutuskan / mengubah koneksi dari jenis yang berbeda, dan bukan yang kita ambil di awal. Kami menemukan jenis koneksi apa yang seharusnya antara atom pertama awal dan atom kedua yang diubah dan memprosesnya sesuai dengan aturan yang sama seperti yang diharapkan semula. Jika dalam hal ini jenis atomnya telah berubah lagi, kita akan menggunakan skema yang sama lagi. Ini secara implisit tersirat di sini,bahwa jenis ikatan baru memiliki sifat yang sama - ikatan tersebut putus dengan panjang yang sama, dll., dan partikel yang terbentuk selama putus sesuai kebutuhan. Karena informasi ini disentuh oleh pengguna, kami mengalihkan tanggung jawab dari program kami kepadanya, dia harus mengatur semuanya dengan benar. Kode:
__global__ void apply_bonds(int iStep, int bndPerBlock, int bndPerThread, cudaMD* md)
{
int def;
int id1, id2; // atom indexes
int old, old_spec2, spec1, spec2, new_spec1, new_spec2; // atom types
int new_bond_type;
int save_lt, need_r, loop; // flags to save lifetime, to need to calculate r^2 and to be in ‘while’ loop
int mnmx; // flag minimum or maximum
int action; // flag: 0 - do nothing, 1 - delete bond, 2 - transform bond
cudaBond *old_bnd, *cur_bnd; // old bond type, current bond type
float dx, dy, dz, r2, r;
float f, eng = 0.0f;
__shared__ float shEng;
#ifdef DEBUG_MODE
int cnt; // count of change spec2 loops
#endif
if (threadIdx.x == 0)
{
shEng = 0.0f;
}
__syncthreads();
int id0 = blockIdx.x * bndPerBlock + threadIdx.x * bndPerThread;
int N = min(id0 + bndPerThread, md->nBond);
int iBnd;
for (iBnd = id0; iBnd < N; iBnd++)
if (md->bonds[iBnd].z) // the bond is not broken
{
// atom indexes
id1 = md->bonds[iBnd].x;
id2 = md->bonds[iBnd].y;
// atom types
spec1 = md->types[id1];
spec2 = md->types[id2];
old_bnd = &(md->bondTypes[md->bonds[iBnd].z]);
cur_bnd = old_bnd;
save_lt = 0;
need_r = 1;
loop = 1;
#ifdef DEBUG_MODE
cnt = 0;
#endif
if ((cur_bnd->spec1 == spec1)&&(cur_bnd->spec2 == spec2))
{
//ok
}
else
if ((cur_bnd->spec1 == spec2) && (cur_bnd->spec2 == spec1))
{
invert_bond(id1, id2, spec1, spec2, &(md->bonds[iBnd]));
//... then ok
}
else // atom types do not correspond to bond types
{
save_lt = 1;
}
// end initial stage
while (loop)
{
if (save_lt)
{
def = md->def_bonds[spec1][spec2];
if (def == 0) // these atom types do not form a bond
{
#ifdef DEBUG_MODE
printf("probably, something goes wrong\n");
#endif
action = 1; // delete
break;
}
else
{
//! change bond type and go on
if (def < 0)
{
invert_bond(id1, id2, spec1, spec2, &(md->bonds[iBnd]));
def = -def;
}
md->bonds[iBnd].z = def;
cur_bnd = &(md->bondTypes[def]);
}
} // end if (save_lt)
// calculate distance (only once)
if (need_r)
{
dx = md->xyz[id1].x - md->xyz[id2].x;
dy = md->xyz[id1].y - md->xyz[id2].y;
dz = md->xyz[id1].z - md->xyz[id2].z;
delta_periodic(dx, dy, dz, md);
r2 = dx * dx + dy * dy + dz * dz;
need_r = 0;
}
action = 0; // 0 - just cultivate bond 1 - delete bond 2 - transform bond
if ((cur_bnd->mxEx) && (r2 > cur_bnd->r2max))
{
mnmx = 1;
if (cur_bnd->new_type[mnmx] == 0) // delete bond
action = 1;
else
action = 2; // modify bond
}
else if ((cur_bnd->mnEx) && (r2 < cur_bnd->r2min))
{
mnmx = 0;
action = 2; // at minimum only bond modification possible
}
// end select action
// try to change atom types (if needed)
if (action)
{
save_lt = 1;
new_spec1 = cur_bnd->new_spec1[mnmx];
new_spec2 = cur_bnd->new_spec2[mnmx];
//the first atom
old = atomicCAS(&(md->types[id1]), spec1, new_spec1);
if (old != spec1)
{
spec1 = old;
spec2 = md->types[id2]; // refresh type of the 2nd atom
// return to begin of the ‘while’ loop
}
else // types[id1] have been changed
{
#ifdef USE_NEWANG // save changes in atom type
atomicCAS(&(md->oldTypes[id1]), -1, spec1);
#endif
old_spec2 = spec2;
while ((old = atomicCAS(&(md->types[id2]), old_spec2, new_spec2)) != old_spec2)
{
//! the worst variant: this thread changes atom 1, other thread changes atom 2
// imagine that we had A-old bond with the same behavior
def = md->def_bonds[spec1][old];
#ifdef DEBUG_MODE
if (def == 0)
{
printf("UBEH[001]: in apply_bonds, change atom types. There are no bond types between Species[%d] and [%d]\n", spec1, old);
break;
}
#endif
if (def < 0) // spec1 -> new_spec2 spec2 -> newSpec1
{
cur_bnd = &(md->bondTypes[-def]);
new_spec2 = cur_bnd->new_spec1[mnmx];
}
else // direct order
{
cur_bnd = &(md->bondTypes[def]);
new_spec2 = cur_bnd->new_spec2[mnmx];
}
old_spec2 = old;
#ifdef DEBUG_MODE
cnt++;
if (cnt > 10)
{
printf("UBEH[002]: too many atempst to change spec2 = %d\n", spec2);
break;
}
#endif
}
#ifdef USE_NEWANG // save changes in atom type
atomicCAS(&(md->oldTypes[id2]), -1, spec2);
#endif
loop = 0;
}
//end change types
} // end if (action)
else
loop = 0; // action == 0, out of cycle
} // end while(loop)
if (action == 2)
{
new_bond_type = cur_bnd->new_type[mnmx];
if (new_bond_type < 0)
{
md->bonds[iBnd].x = id2;
md->bonds[iBnd].y = id1;
new_bond_type = -new_bond_type;
}
md->bonds[iBnd].z = new_bond_type;
cur_bnd = &(md->bondTypes[new_bond_type]);
}
// perform calculations and save mean bond length
if (action != 1) // not delete
{
r = sqrt(r2);
f = cur_bnd->force_eng(r2, r, eng, cur_bnd);
atomicAdd(&(md->frs[id1].x), f * dx);
atomicAdd(&(md->frs[id2].x), -f * dx);
atomicAdd(&(md->frs[id1].y), f * dy);
atomicAdd(&(md->frs[id2].y), -f * dy);
atomicAdd(&(md->frs[id1].z), f * dz);
atomicAdd(&(md->frs[id2].z), -f * dz);
atomicAdd(&(cur_bnd->rSumm), r);
atomicAdd(&(cur_bnd->rCount), 1);
}
else //delete bond
{
// decrease the number of bonds for atoms
atomicSub(&(md->nbonds[id1]), 1);
atomicSub(&(md->nbonds[id2]), 1);
md->bonds[iBnd].z = 0;
// change parents
exclude_parents(id1, id2, md);
}
if (save_lt)
{
keep_bndlifetime(iStep, &(md->bonds[iBnd]), old_bnd);
if (action != 1) // not delete
atomicAdd(&(cur_bnd->count), 1);
atomicSub(&(old_bnd->count), 1);
}
} // end main loop
// split energy to shared and then to global memory
atomicAdd(&shEng, eng);
__syncthreads();
if (threadIdx.x == 0)
atomicAdd(&(md->engBond), shEng);
}
Dalam kode, saya menggunakan arahan preprocessor untuk mengaktifkan pemeriksaan untuk situasi yang mungkin timbul karena pengawasan pengguna. Anda dapat mematikannya untuk mempercepat kinerja. Fungsi tersebut mengimplementasikan skema di atas, tetapi dibungkus dalam satu loop lagi yang berjalan melalui berbagai link yang menjadi tanggung jawab thread ini. Selanjutnya identifier jenis ikatan bisa negatif, ini berarti urutan atom dalam ikatan harus dibalik (misalnya ikatan OH dan HO adalah ikatan yang sama, tetapi dalam algoritma urutannya penting, untuk menunjukkannya, saya menggunakan indeks dengan kebalikannya tanda), fungsi invert_bond membuatnya terlalu sepele untuk dijelaskan. Fungsi Delta_periodicmenerapkan kondisi batas periodik untuk mengkoordinasikan perbedaan. Jika kita perlu mengubah tidak hanya ikatan, tetapi juga sudut ikatan (direktif USE_NEWANG ), maka kita perlu menandai atom yang telah kita ubah jenisnya (lebih lanjut nanti). Untuk mengecualikan pengikatan ulang atom yang sama dengan ikatan, larik induk menyimpan indeks salah satu atom yang terkait dengan data (jaring pengaman ini tidak berfungsi di semua kasus, tetapi untuk tambang itu sudah cukup). Jika kita memutuskan beberapa jenis koneksi, maka kita perlu menghapus indeks atom yang sesuai dari array orang tua, ini dilakukan oleh fungsi exclude_parents :
__device__ void exclude_parents(int id1, int id2, cudaMD* md)
// exclude id1 and id2 from parents of each other (if they are)
// and seek other parents if able
{
// flags to clear parent
int clear_1 = 0;
int clear_2 = 0;
int i, flag;
if (md->parents[id1] == id2)
clear_1 = 1;
if (md->parents[id2] == id1)
clear_2 = 1;
i = 0;
while ((i < md->nBond) && (clear_1 || clear_2))
{
if (md->bonds[i].z != 0)
{
flag = 0;
if (clear_1)
{
if (md->bonds[i].x == id1)
{
md->parents[id1] = md->bonds[i].y;
flag = 1;
}
else if (md->bonds[i].y == id1)
{
md->parents[id1] = md->bonds[i].y;
flag = 1;
}
if (flag)
{
clear_1 = 0;
i++;
continue;
}
}
if (clear_2)
{
if (md->bonds[i].x == id2)
{
md->parents[id2] = md->bonds[i].y;
flag = 1;
}
else if (md->bonds[i].y == id2)
{
md->parents[id2] = md->bonds[i].y;
flag = 1;
}
if (flag)
{
clear_2 = 0;
i++;
continue;
}
}
}
i++;
}
// be on the safe side
if (clear_1)
md->parents[id1] = -1;
if (clear_2)
md->parents[id2] = -1;
}
Sayangnya, fungsinya berjalan melalui seluruh rangkaian tautan. Kami belajar cara memproses dan menghapus tautan, sekarang kami perlu belajar cara membuatnya. Fungsi berikut menandai atom-atom yang cocok untuk membentuk ikatan kimia:
__device__ void try_to_bind(float r2, int id1, int id2, int spec1, int spec2, cudaMD *md)
{
int r2Int; // (int)r2 * const
// save parents to exclude re-linking
if (md->parents[id1] == id2)
return;
if (md->parents[id2] == id1)
return;
if (md->bindBonds[spec1][spec2] != 0)
{
if (r2 < md->bindR2[spec1][spec2])
{
r2Int = (int)(r2 * 100);
if (atomicMin(&(md->r2Min[id1]), r2Int) > r2Int) // replace was sucessfull
{
md->neighToBind[id1] = id2 + 1; // as 0 is reserved for no neighbour
md->canBind[id1] = 1;
}
// similar for the second atom
if (atomicMin(&(md->r2Min[id2]), r2Int) > r2Int) // replace was sucessfull
{
md->neighToBind[id2] = id1 + 1; // as 0 is reserved for no bind
md->canBind[id2] = 1;
}
}
}
}
Matriks bindBonds menyimpan informasi tentang apakah jenis atom ini dapat membentuk ikatan, dan jika demikian, yang mana. Matriks bindR2 menyimpan jarak maksimum antar atom yang diperlukan untuk pengikatan. Jika semua kondisi mendukung, maka kami memeriksa apakah atom tetangga lain cocok untuk ikatan, tetapi lebih dekat.
Informasi tentang jarak terdekat ke tetangga disimpan dalam larik r2Min (untuk kenyamanan, larik berjenis int dan nilainya diubah dengannya dengan perkalian dengan konstanta, 100). Jika tetangga yang terdeteksi adalah yang terdekat, maka kita mengingat indeksnya dalam larik neighToBind dan menyetel bendera canBind... Ada bahaya nyata bahwa sementara kami melanjutkan untuk memperbarui indeks, utas lain menimpa nilai minimum, tetapi ini tidak terlalu penting. Sebaiknya panggil fungsi ini dalam fungsi yang melintasi pasangan atom, misalnya, cell_list atau all_pair , yang dijelaskan di bagian pertama . Pengikatan itu sendiri:
__global__ void create_bonds(int iStep, int atPerBlock, int atPerThread, cudaMD* md)
// connect atoms which are selected to form bonds
{
int id1, id2, nei; // neighbour index
int btype, bind; // bond type index and bond index
cudaBond* bnd;
int spec1, spec2; // species indexes
int id0 = blockIdx.x * atPerBlock + threadIdx.x * atPerThread;
int N = min(id0 + atPerThread, md->nAt);
int iat;
for (iat = id0; iat < N; iat++)
{
nei = md->neighToBind[iat];
if (nei) // neighbour exists
{
nei--; // (nei = spec_index + 1)
if (iat < nei)
{
id1 = iat;
id2 = nei;
}
else
{
id1 = nei;
id2 = iat;
}
// try to lock the first atom
if (atomicCAS(&(md->canBind[id1]), 1, 0) == 0) // the atom is already used
continue;
// try to lock the second atom
if (atomicCAS(&(md->canBind[id2]), 1, 0) == 0) // the atom is already used
{
// unlock the first one back
atomicExch(&(md->canBind[id1]), 1);
continue;
}
// create bond iat-nei
bind = atomicAdd(&(md->nBond), 1);
#ifdef DEBUG_MODE
if (bind >= md->mxBond)
{
printf("UBEH[003]: Exceed maximal number of bonds, %d\n", md->mxBond);
}
#endif
spec1 = md->types[id1];
spec2 = md->types[id2];
#ifdef USE_NEWANG // save that we have changed atom type
atomicCAS(&(md->oldTypes[id1]), -1, spec1);
atomicCAS(&(md->oldTypes[id2]), -1, spec2);
#endif
btype = md->bindBonds[spec1][spec2];
if (btype < 0)
{
// invert atoms order
md->bonds[bind].x = id2;
md->bonds[bind].y = id1;
md->bonds[bind].z = -btype;
bnd = &(md->bondTypes[-btype]);
// change atom types according the formed bond
md->types[id1] = bnd->spec2;
md->types[id2] = bnd->spec1;
}
else
{
md->bonds[bind].x = id1;
md->bonds[bind].y = id2;
md->bonds[bind].z = btype;
bnd = &(md->bondTypes[btype]);
// change atom types according the formed bond
md->types[id1] = bnd->spec1;
md->types[id2] = bnd->spec2;
}
atomicAdd((&bnd->count), 1);
md->bonds[bind].w = iStep; // keep time of the bond creation for lifetime calculation
atomicAdd(&(md->nbonds[id1]), 1);
atomicAdd(&(md->nbonds[id2]), 1);
// replace parents if none:
atomicCAS(&(md->parents[id1]), -1, id2);
atomicCAS(&(md->parents[id2]), -1, id1);
}
}
// end loop by atoms
}
Fungsi tersebut memblokir satu atom, kemudian yang kedua dan, jika berhasil memblokir keduanya, membuat koneksi di antara mereka. Di awal fungsi, indeks atom diurutkan untuk mengecualikan situasi ketika satu utas memblokir atom pertama dalam suatu pasangan, dan utas lainnya memblokir atom kedua dalam pasangan yang sama, kedua utas berhasil melewati pemeriksaan pertama dan gagal pada yang kedua, sebagai akibatnya tidak satu pun dari koneksi tidak membuatnya. Dan terakhir, kami perlu menghapus tautan yang kami tandai untuk dihapus di fungsi apply_bonds :
__global__ void clear_bonds(cudaMD* md)
// clear bonds with .z == 0
{
int i = 0;
int j = md->nBond - 1;
while (i < j)
{
while ((md->bonds[j].z == 0) && (j > i))
j--;
while ((md->bonds[i].z != 0) && (i < j))
i++;
if (i < j)
{
md->bonds[i] = md->bonds[j];
md->bonds[j].z = 0;
i++;
j--;
}
}
if ((i == j) && (md->bonds[i].z == 0))
md->nBond = j;
else
md->nBond = j + 1;
}
Kami hanya memindahkan tautan "dibatalkan" ke akhir larik dan mengurangi jumlah tautan sebenarnya. Sayangnya, kodenya berseri, tetapi saya tidak yakin bahwa memparalelkannya akan memiliki efek yang nyata. Fungsi yang menghitung energi ikat aktual dan gaya pada atom, yang ditunjukkan oleh medan gaya_eng dari struktur cudaBond , masih diabaikan , tetapi sepenuhnya analog dengan fungsi potensial pasangan yang dijelaskan di bagian pertama.
Sudut valensi
Dengan sudut valensi, saya akan memperkenalkan beberapa asumsi untuk membuat algoritme dan fungsi lebih mudah, dan sebagai hasilnya, mereka akan lebih sederhana daripada untuk ikatan valensi. Pertama, parameter sudut ikatan harus bergantung pada ketiga atom, tetapi di sini kita akan mengasumsikan bahwa jenis sudut ikatan secara eksklusif menentukan atom pada puncaknya. Saya mengusulkan algoritma paling sederhana untuk membentuk / menghapus sudut: setiap kali kita mengubah jenis atom, kita mengingat fakta ini di dalam array oldTypes [] . Besar kecilnya array sama dengan jumlah atom, awalnya diisi dengan -1. Jika suatu fungsi mengubah jenis atom, ia menggantikan -1 dengan indeks jenis aslinya. Untuk semua atom yang telah mengubah jenisnya, hilangkan semua sudut ikatan dan hilangkan semua ikatan atom ini untuk menjumlahkan sudut yang sesuai:
__global__ void refresh_angles(int iStep, int atPerBlock, int atPerThread, cudaMD *md)
// delete old angles and create new ones for atoms which change their type
{
int i, j, n, t, ang;
int nei[8]; // bonded neighbors of given atom
int cnt;
int id0 = blockIdx.x * atPerBlock + threadIdx.x * atPerThread;
int N = min(id0 + atPerThread, md->nAt);
int iat;
for (iat = id0; iat < N; iat++)
if (md->oldTypes[iat] != -1)
{
i = 0;
n = md->nangles[iat];
while (n && (i < md->nAngle))
{
if (md->angles[i].w)
if (md->angles[i].x == iat)
{
n--;
md->angles[i].w = 0;
}
i++;
}
// create new angles
t = md->specAngles[md->types[iat]]; // get type of angle, which formed by current atom type
if (t && (md->nbonds[iat] > 1)) // atom type supports angle creating and number of bonds is enough
{
// search of neighbors by bonds
i = 0; cnt = 0;
n = md->nbonds[iat];
while (n && (i < md->nBond))
{
if (md->bonds[i].z) // if bond isn't deleted
{
if (md->bonds[i].x == iat)
{
nei[cnt] = md->bonds[i].y;
cnt++;
n--;
}
else if (md->bonds[i].y == iat)
{
nei[cnt] = md->bonds[i].x;
cnt++;
n--;
}
}
i++;
}
// add new angles based on found neighbors:
for (i = 0; i < cnt-1; i++)
for (j = i + 1; j < cnt; j++)
{
ang = atomicAdd(&(md->nAngle), 1);
md->angles[ang].x = iat;
md->angles[ang].y = nei[i];
md->angles[ang].z = nei[j];
md->angles[ang].w = t;
}
n = (cnt * (cnt - 1)) / 2;
}
md->nangles[iat] = n;
// reset flag
md->oldTypes[iat] = -1;
}
}
Array specAngles berisi pengenal sudut ikatan yang sesuai dengan jenis atom yang diberikan. Fungsi berikut memanggil kalkulasi energi dan gaya untuk semua sudut:
__global__ void apply_angles(int iStep, int angPerBlock, int angPerThread, cudaMD* md)
// apply valence angle potentials
{
cudaAngle* ang;
// energies of angle potential
float eng;
__shared__ float shEng;
if (threadIdx.x == 0)
shEng = 0.0f;
__syncthreads();
int id0 = blockIdx.x * angPerBlock + threadIdx.x * angPerThread;
int N = min(id0 + angPerThread, md->nAngle);
int i;
for (i = id0; i < N; i++)
if (md->angles[i].w)
{
ang = &(md->angleTypes[md->angles[i].w]);
ang->force_eng(&(md->angles[i]), ang, md, eng);
}
// split energy to shared and then to global memory
atomicAdd(&shEng, eng);
__syncthreads();
if (threadIdx.x == 0)
atomicAdd(&(md->engAngl), shEng);
}Nah, misalnya, potensi sudut seperti itu, memberikan fungsi kosinus harmonis, yang mungkin menunjukkan medan gaya_eng struktur cudaAngle :
__device__ void angle_hcos(int4* angle, cudaAngle* type, cudaMD* md, float& eng)
// harmonic cosine valent angle potential:
// U = k / 2 * (cos(th)-cos(th0))^
{
float k = type->p0;
float cos0 = type->p1;
// indexes of central atom and ligands:
int c = angle->x;
int l1 = angle->y;
int l2 = angle->z;
// vector ij
float xij = md->xyz[l1].x - md->xyz[c].x;
float yij = md->xyz[l1].y - md->xyz[c].y;
float zij = md->xyz[l1].z - md->xyz[c].z;
delta_periodic(xij, yij, zij, md);
float r2ij = xij * xij + yij * yij + zij * zij;
float rij = sqrt(r2ij);
// vector ik
float xik = md->xyz[l2].x - md->xyz[c].x;
float yik = md->xyz[l2].y - md->xyz[c].y;
float zik = md->xyz[l2].z - md->xyz[c].z;
delta_periodic(xik, yik, zik, md);
float r2ik = xik * xik + yik * yik + zik * zik;
float rik = sqrt(r2ik);
float cos_th = (xij * xik + yij * yik + zij * zik) / rij / rik;
float dCos = cos_th - cos0; // delta cosinus
float c1 = -k * dCos;
float c2 = 1.0 / rij / rik;
atomicAdd(&(md->frs[c].x), -c1 * (xik * c2 + xij * c2 - cos_th * (xij / r2ij + xik / r2ik)));
atomicAdd(&(md->frs[c].y), -c1 * (yik * c2 + yij * c2 - cos_th * (yij / r2ij + yik / r2ik)));
atomicAdd(&(md->frs[c].z), -c1 * (zik * c2 + zij * c2 - cos_th * (zij / r2ij + zik / r2ik)));
atomicAdd(&(md->frs[l1].x), c1 * (xik * c2 - cos_th * xij / r2ij));
atomicAdd(&(md->frs[l1].y), c1 * (yik * c2 - cos_th * yij / r2ij));
atomicAdd(&(md->frs[l1].z), c1 * (zik * c2 - cos_th * zij / r2ij));
atomicAdd(&(md->frs[l2].x), c1 * (xij * c2 - cos_th * xik / r2ik));
atomicAdd(&(md->frs[l2].y), c1 * (yij * c2 - cos_th * yik / r2ik));
atomicAdd(&(md->frs[l2].z), c1 * (zij * c2 - cos_th * zik / r2ik));
eng += 0.5 * k * dCos * dCos;
}
Saya tidak akan memberikan fungsi untuk menghapus sudut "nullified", ini tidak berbeda secara fundamental dari clear_bonds .
Contoh dari
Tanpa berpura-pura akurat, saya mencoba menggambarkan perakitan molekul air dari ion individu. Potensial berpasangan ditetapkan sewenang-wenang berupa potensial Buckingham, kemudian ditambahkan kemampuan membuat ikatan berupa potensial harmonik, dengan jarak kesetimbangan sama dengan panjang ikatan HO dalam air, 0,96 Å. Selain itu, ketika proton kedua terikat dengan oksigen, sudut ikatan dengan puncak oksigen ditambahkan. Setelah 100'000 langkah, molekul pertama muncul dari ion yang tersebar secara acak. Gambar tersebut menunjukkan konfigurasi awal (kiri) dan terakhir (kanan):

Anda dapat membuat percobaan seperti ini: biarkan pada awalnya semua atom sama, tetapi ketika mereka bersebelahan, mereka membentuk ikatan. Biarkan atom terikat membentuk ikatan lain baik dengan atom bebas atau dengan molekul terikat serupa lainnya. Hasilnya, kita mendapatkan semacam organisasi mandiri, di mana atom-atom berbaris dalam rantai:

Komentar terakhir
- Di sini kami hanya menggunakan satu kriteria untuk jarak ikatan, meskipun pada prinsipnya mungkin ada kriteria lain, misalnya, energi sistem. Pada kenyataannya, ketika ikatan kimia terbentuk, biasanya energi dilepaskan dalam bentuk panas. Ini tidak diperhitungkan di sini, tetapi Anda dapat mencoba menerapkannya, misalnya, mengubah kecepatan partikel.
- Interaksi antar partikel melalui potensi ikatan kimia tidak meniadakan fakta bahwa partikel masih dapat berinteraksi melalui potensi pasangan antarmolekul dan interaksi Coulomb. Tentu saja dimungkinkan untuk tidak menghitung interaksi antarmolekul untuk atom terikat, tetapi ini, dalam kasus umum, membutuhkan pemeriksaan yang lama. Oleh karena itu, lebih mudah untuk mengatur potensial ikatan kimia sedemikian rupa sehingga jumlah dengan potensial lain memberikan fungsi yang diinginkan.
- Implementasi paralel dari pengikatan partikel tidak hanya memberikan peningkatan kecepatan, tetapi bahkan terlihat lebih realistis, karena partikel-partikel tersebut bersaing satu sama lain.
Nah, ada beberapa proyek di Habré yang sangat mirip dengan yang ini: